
INTRODUCTION
|
Not a Medical Device… Until MDR Says You Are! (Hello Annex XVI)
|
|---|
|
Devices without an intended medical purpose can still fall under EU MDR 2017/745 if they are listed in Annex XVI. The idea is simple: even if you’re not claiming “medical use,” these products can interact with the body in similar ways to medical devices and can create similar risks so the EU regulates them like medical devices to protect users. Annex XVI covers 6 groups of products, like cosmetic (non-corrective) contact lenses, dermal fillers, body-contouring implants, fat reduction equipment (e.g., liposuction/lipolysis), high-intensity radiation devices used for aesthetic purposes (lasers/IPL for skin/hair/tattoo work), and brain stimulation devices used for wellness/enhancement. Because they can involve tissue contact, injections, energy delivery, or anatomical modification, manufacturers must prove safety and performance even without a medical “benefit.” For these products, manufacturers must follow most key MDR duties: set up a QMS (Article 10), appoint a PRRC (Article 15), prepare technical documentation, meet GSPRs (Annex I), do the right conformity assessment + CE marking, and run PMS/PMCF. Also, Annex XVI products must comply with the Common Specifications (EU) 2022/2346, which give detailed requirements on risk management, labelling, performance evidence, and post-market activities. Some devices are also reclassified under EU 2022/2347 (e.g., many aesthetic energy devices become Class IIb; brain stimulation penetrating the cranium becomes Class III), which affects whether a Notified Body is required. Finally, manufacturers should check transitional timelines: Common Specifications apply from 22 June 2023, and certain transitional deadlines have been extended (some pathways now go up to 31 Dec 2028/31 Dec 2029, depending on conditions like being lawfully marketed before 22 June 2023 and avoiding significant changes). The practical workflow is: confirm Annex XVI applicability → classify correctly → build/upgrade QMS + PRRC |
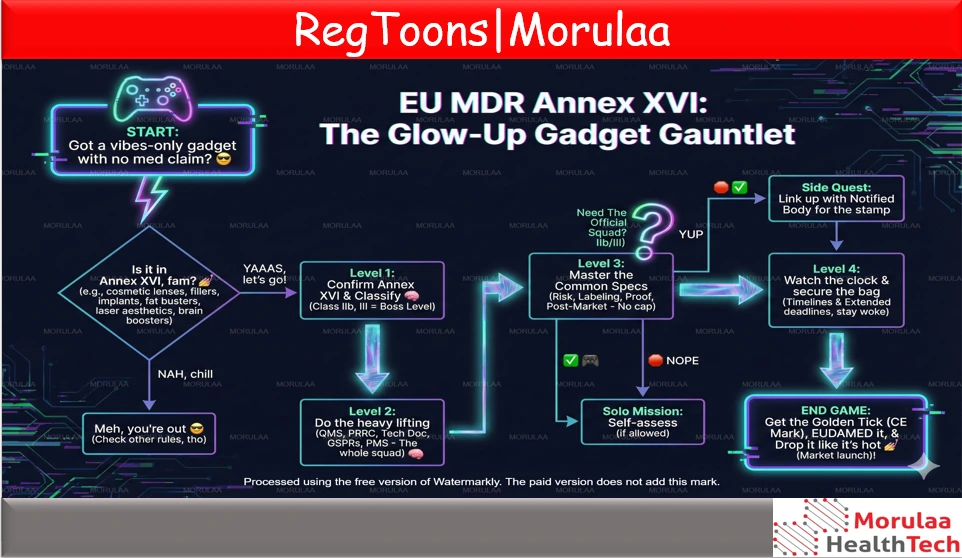
WHAT ARE “DEVICES WITHOUT AN INTENDED MEDICAL PURPOSE”?
Annex XVI of the MDR identifies specific groups of products that, despite lacking a medical purpose, interact with the human body in ways analogous to medical devices.The European Commission concluded that because such products may present risks similar to those of medical devices (e.g., tissue modification, administration of energy, body contact), they should be regulated under the MDR framework.Typical examples include:
- Non‑corrective coloured contact lenses (for cosmetic effect).
- Body contour‑modifying implants or subcutaneous filling substances.
- Equipment for adipose tissue reduction (e.g., liposuction devices) and high‑intensity electromagnetic radiation devices (e.g., lasers, IPL) for aesthetic treatment.
- Non‑invasive brain stimulation devices for non‑medical purposes (e.g., wellness or enhancement).
Even though they are not marketed for diagnosis or treatment of disease, their interaction with the body gives rise to health and safety considerations.
WHY DOES MDR APPLY TO THESE DEVICES?
The MDR’s main trigger is “intended medical purpose” but Annex XVI functions as an exception: it brings in those devices without a medical intended purpose but with comparable risk/technology characteristics. The reasoning is grounded in the risk to the person using them. For example:
- Contact with tissues, subcutaneous injection, surgical access, administration of energy or modification of anatomy all carry intrinsic risk.
- The regulation emphasises that even in absence of a therapeutic benefit, safety and performance must be demonstrated.
Therefore, manufacturers must treat Annex XVI devices in much the same way as medical devices with respect to regulatory obligations.
ANNEX XVI PRODUCT GROUPS AND EXAMPLES
The Groups of Products Without an Intended Medical Purpose as referred to in Article 1(2) of the Medical Device Regulation (EU) 2017/745:
S. No. | Product Group | Description |
1 | Contact Lenses or Similar | Contact lenses or other items intended to be introduced into or onto the eye. |
2 | Anatomical Modification Implants | Products intended to be totally or partially introduced into the human body through surgically invasive means to modify anatomy or for fixation of body parts (excluding tattooing products and piercings). |
3 | Dermal Fillers | Substances, combinations of substances, or items for facial or other dermal or mucous membrane filling via subcutaneous, submucous, or intradermal injection or other means (excluding tattooing). |
4 | FatReduction Equipment | Equipment intended to reduce, remove or destroy adipose tissue, e.g., for liposuction, lipolysis, or lipoplasty. |
5 | High-Intensity Radiation Devices | Devices emitting high-intensity electromagnetic radiation (infra-red, visible, UV), such as lasers or intense pulsed light, for skin resurfacing, tattoo or hair removal, or other skin treatments. |
6 | Brain Stimulation Equipment | Equipment for brain stimulation using electrical currents or magnetic/electromagnetic fields that penetrate the cranium to modify neuronal activity. |
CLASSIFICATION & RECLASSIFICATION OF ANNEX XVI DEVICES
Although classification under MDR is typically based on intended medical purpose, Annex XVI devices follow specific classification logic.
- The general classification rules of Annex VIII apply, with modification for active devices without intended medical purpose.
- Under Regulation (EU) 2022/2347 certain active devices in Annex XVI are reclassified:
- Equipment emitting high‑intensity electromagnetic radiation for skin treatment is Class IIb unless only intended for hair removal, then Class IIa.
- Equipment for adipose tissue reduction is Class IIb
- Equipment for brain stimulation (penetrating the cranium) is Class III
- Equipment emitting high‑intensity electromagnetic radiation for skin treatment is Class IIb unless only intended for hair removal, then Class IIa.
Manufacturers must therefore determine the correct risk class, which dictates the level of conformity assessment and Notified Body involvement.
KEY MDR REQUIREMENTS APPLICABLE TO ANNEX XVI DEVICES
Manufacturers of Annex XVI devices must comply with the majority of MDR obligations, including:
- Establishment of a Quality Management System (QMS) appropriate to the device risk class. MDR Article 10(9) applies.
- Appointment of a Person Responsible for Regulatory Compliance (PRRC) as per MDR Article 15.
- Technical documentation, including demonstration of safety and performance (though device does not need a clinical benefit if it has no medical purpose).
- Compliance with the General Safety & Performance Requirements (GSPRs) in Annex I of the MDR, including GSPR #9 which is specifically oriented to risk versus benefit for devices without medical purpose.
- For these devices, instead of clinical benefit, manufacturers must demonstrate performance and safety (via clinical data where needed, post‑market surveillance, post‑market clinical follow‑up).
- Conformity assessment and CE‑marking consistent with the assigned classification and Notified Body requirement.
Additionally, compliance with the Commission Implementing Regulation (EU) 2022/2346 (CS) is required: these Common Specifications set out detailed requirements for Annex XVI devices including risk management, labelling, performance and post‑market activities.
TRANSITIONAL PROVISIONS & DEADLINES
Manufacturers should be aware of key dates :
- The CS under Regulation 2022/2346 apply from 22 June 2023 for devices not requiring Notified Body involvement.
- For devices requiring Notified Body involvement, transitional deadlines depend on whether a clinical investigation is planned:
- Originally 22 June 2025 for devices without trial, now extended to 31 Dec 2028.
- For devices with clinical investigation plan: originally 22 June 2028, now extended to 31 Dec 2029.
- Originally 22 June 2025 for devices without trial, now extended to 31 Dec 2028.
- To benefit from those transitional periods the device must have been lawfully marketed before 22 June 2023 and not subject to significant change in design or intended purpose.
Manufacturers should check whether they qualify for transitional provisions and begin conformity activities accordingly.
PRACTICAL STEPS FOR MANUFACTURER COMPLIANCE
- Identify whether your product falls under Annex XVI: Review the list of groups and check your device’s intended purpose, technology and risk profile.
- Classify the device correctly: Determine whether your device is covered by MDR, whether it is active, and check if Regulation 2022/2347 applies.
- Set up or update your QMS and assign PRRC: Ensure you have QMS in place per MDR Article 10 and a qualified PRRC per Article 15.
- Prepare technical documentation and conformity assessment plans: Based on classification, decide what conformity route applies, involve a Notified Body if required.
- Demonstrate safety and performance: For devices without intended medical purpose, demonstrate performance and safety via relevant data; ensure alignment with CS under Regulation 2022/2346.
- Conduct post‑market surveillance and follow‑up: Establish systems for monitoring once the device is on the market, gather post‑market data, update documentation regularly.
- Monitor regulatory updates and amendments: Annex XVI may be expanded via delegated acts; CS and transitional timelines may also evolve.
- Register the device and economic operators: Ensure registration in the relevant EU database (such as EUDAMED) and fulfil operator obligations under MDR.
CONCLUSION
For manufacturers of devices without an intended medical purpose, the regulatory landscape under the MDR is demanding but clearly defined. By recognizing that your products may fall under Annex XVI, understanding the correct classification, fulfilling the QMS and conformity assessment requirements, and ensuring alignment with the Common Specifications, you can achieve compliance and market your products safely in the EU. Staying abreast of transitional deadlines and regulatory adjustments is essential to avoid non‑compliance risks. The guiding principle remains: although these devices do not bear a medical purpose, their safety and performance are of critical importance.
HOW MORULAA CAN HELP
Morulaa supports manufacturers of Annex XVI devices in achieving MDR compliance by offering end-to-end regulatory assistance. We help set up or upgrade your Quality Management System, prepare complete technical documentation, and guide you in demonstrating safety and performance through appropriate clinical evaluation strategies. Our team ensures your device aligns with Common Specifications (EU) 2022/2346, manages classification and conformity assessment with Notified Bodies, and handles EUDAMED registrations.
FAQs
When are the CS applicable to Annex XVI products?
The CS are applicable to Annex XVI products from 22 June 2023. Certain provisions for products that are covered by a certificate issued by a notified body in accordance with the MDD are applicable from 22 December 2022 (see question 3).
When is the MDR applicable to Annex XVI products?
The MDR is applicable to Annex XVI products from 22 June 2023 , which is the date of application of the CS.
What is the transitional period set out in the MDR for Annex XVI products covered by an MDD certificate? Are there conditions to be satisfied?
The period ends on 31 December 2027 for higher risk products (all class III devices and class IIb implantable devices except sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors) and on 31 December 2028 for lower risk products (all other classes and devices excluded from the higher risk group).
Not all products covered by an MDD certificate are eligible to benefit from the transitional provisions established by the MDR. The following conditions apply:
a) If the MDD certificate was issued from 25 May 2017, was still valid on 26 May 2021, have not been withdrawn afterwards and expired before 20 March 2023, the covered products can benefit from the MDR transitional provisions only if one of the conditions established in Article 120(2), second subparagraph, points a) or b) is met;
b) If the MDD certificate was issued from 25 May 2017, was still valid on 26 May 2021, has not been withdrawn afterwards and has not expired before 20 March 2023 the covered products can benefit from the MDR transitional provisions. The paragraphs (3c), (3d) and (3e) of Article 120 of the MDR establish additional conditions and requirements that must be fulfilled to benefit and continue to benefit from the transitional provisions. Additional information on those specific conditions is provided in the questions and answers document on the implementation of Regulation (EU) 2023/60715 .
Can notified bodies issue certificates for Annex XVI products under the MDR during the transitional periods?
Yes, they can. Notified bodies can issue certificates for Annex XVI products under the MDR during the transitional periods starting from 22 June 2023 (the date of application of the CS). Before that date, the MDR was not applicable to Annex XVI products, consequently notified bodies could not issue any certificate.