
|
WHY ISO 13485 ALONE IS NOT ENOUGH UNDER EU MDR
|
|---|
|
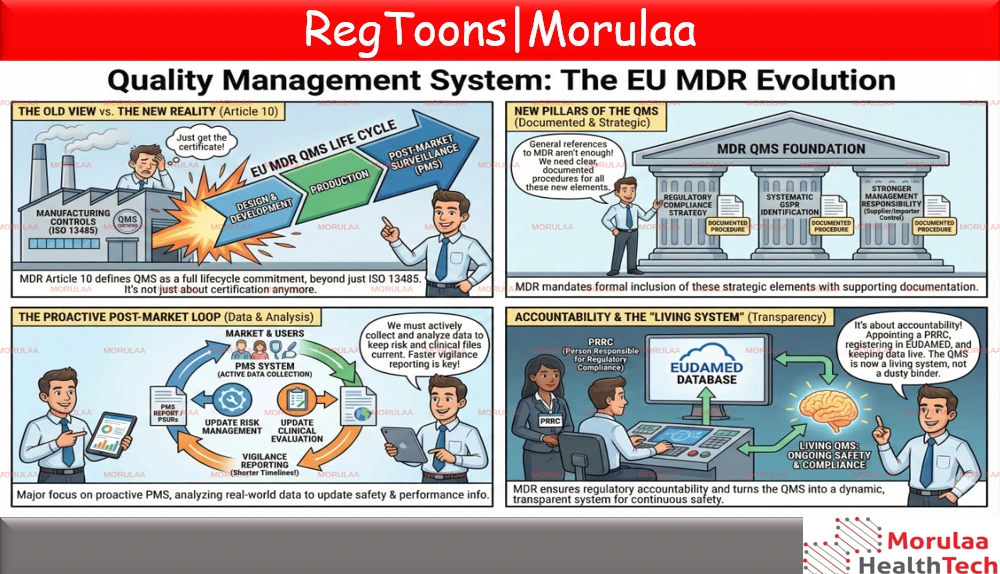
Under the EU Medical Device Regulation (EU MDR 2017/745), a Quality Management System (QMS) is no longer just about manufacturing controls. While ISO 13485:2016 is still required, it is not sufficient on its own. The MDR legally defines what must be included in your QMS, especially under Article 10, which sets out the general obligations of manufacturers. In simple terms, your QMS must now cover the entire life cycle of the device from design and production to post-market monitoring. |
INTRODUCTION
If you are transitioning from the old Medical Device Directive (MDD) to the Medical Device Regulation (EU) 2017/745 (MDR), you have likely heard the warning echoing through the industry: “ISO 13485 is no longer enough.” While ISO 13485:2016 remains the gold standard framework for a Quality Management System (QMS), the EU MDR legislates specific content that must live within that framework. The regulation effectively moves your QMS from a static manufacturing control system to a dynamic, lifecycle driven ecosystem. This guide breaks down exactly how QMS satisfies the “General obligations of manufacturers” outlined in Article 10 and the conformity assessment procedures in Annex IX.
WHAT A QMS DOES IN MEDICAL DEVICES
The Legal Core: Article 10(9)
Under the old MDD, quality requirements were somewhat scattered. The MDR centralizes them in Article 10, Paragraph 9. This single paragraph acts as the “constitution” for your quality system.
It explicitly states that your QMS must be proportionate to the risk class and type of device. Crucially, it lists specific aspects that must be addressed. If your current Quality Manual does not explicitly point to procedures for these items, you are technically non-compliant.
The “Must-Address” List:
- Strategy for Regulatory Compliance: You need a documented procedure ensuring you stay up-to-date with conformity assessment procedures and device changes.
- GSPRs (General Safety and Performance Requirements): Replacing the old “Essential Requirements,” your QMS must systematically identify applicable GSPRs from Annex I.
- Management Responsibility: This is no longer just a high-level commitment; it requires specific resource management procedures, including the selection and control of suppliers and sub-contractors.
Pro Tip: Don’t just “reference” the MDR in your manual. Create a traceability matrix that links every sub-point of Article 10(9) to a specific SOP in your system.
The New “Must-Have” SOPs
To bridge the gap between ISO 13485 and the MDR, you need to develop or significantly update specific Standard Operating Procedures (SOPs).
A. The Person Responsible for Regulatory Compliance (PRRC)
(Regulation Reference: Article 15)
You can no longer assign regulatory duties to a generic quality manager. Your QMS must designate a PRRC.
- Requirement: This person must have specific qualifications (university degree + 1 year exp, or 4 years exp).
- Responsibility: They are personally responsible for ensuring technical documentation is drawn up and PMS obligations are met.
- QMS Action: Create a dedicated job description or appointment letter within your QMS that references Article 15 requirements directly.
B. Economic Operators & Supply Chain Control
(Regulation Reference: Articles 11, 13, 14, and 25)
The MDR views the supply chain as a critical safety valve. Your QMS must extend its reach beyond your factory walls.
- Importers and Distributors: You must verify they meet their own Article 13 and 14 requirements.
- QMS Action: Update your Supplier Evaluation and Purchasing procedures. You need specific checklists to verify that your importers and distributors are compliant. You cannot legally ship to a non-compliant distributor.
C. The Post-Market Surveillance (PMS)
(Regulation Reference: Article 83 and Annex III)
This is arguably the biggest shift. PMS is no longer a “reactive” complaint handling process; it is a “proactive” system.
- The Plan: You need a PMS Plan (Article 84) that defines exactly how and when you will collect data.
- The Report:
- Class I: Post-Market Surveillance Report (PMSR) – Article 85.
- Class IIa, IIb, III: Periodic Safety Update Report (PSUR) – Article 86.
- Class I: Post-Market Surveillance Report (PMSR) – Article 85.
- QMS Action: Your QMS must link PMS data directly back to your Risk Management (Annex I, Chapter 1) and Clinical Evaluation (Article 61). If PMS data shows a new side effect, your Risk Assessment must be updated immediately.
D. Clinical Evaluation & PMCF
(Regulation Reference: Article 61 and Annex XIV)
Clinical evaluation is now a continuous process.
- PMCF: Post-Market Clinical Follow-up is mandatory unless explicitly justified. Your QMS needs a specific procedure for designing and maintaining PMCF studies.
- QMS Action: Ensure your Design and Development procedures don’t “end” at product launch. They must cycle back into Clinical Evaluation Reports (CERs) that are updated annually for high-risk devices.
Vigilance and Timeline Changes
(Regulation Reference: Articles 87-89)
The timelines for reporting serious incidents have tightened, and your SOPs must reflect this exactly to avoid audit findings.
- Serious Incidents: Must be reported no later than 15 days (previously often 30).
- Public Health Threat: 2 days.
- Death/Unanticipated Serious Deterioration: 10 days.
Audit Warning: Auditors will check your Vigilance SOP for these specific numbers. If you still have “30 days” listed, you will receive a non-conformity.
EUDAMED Integration
(Regulation Reference: Article 33)
The European Database on Medical Devices (EUDAMED) is the digital backbone of the MDR. Your QMS requires a procedure for:
- UDI-DI Registration: Assigning and managing Unique Device Identifiers (Article 27).
- Data Uploads: Ensuring the Single Registration Number (SRN) is obtained and data is uploaded correctly.
- Maintenance: Keeping EUDAMED data synchronized with your internal changes.
SUMMARY CHECKLIST FOR THE QUALITY MANAGER
If you are preparing for a Notified Body audit, use this checklist to gauge your readiness:
- Gap Analysis: Have we mapped Article 10(9) requirements to our ISO 13485 clauses?
- PRRC: Is the Person Responsible for Regulatory Compliance appointed in writing?
- PMS Plan: Do we have a proactive PMS plan for every device family?
- Vigilance Timelines: Are the 15/10/2 day reporting windows strictly defined in our SOPs?
- Supply Chain: Have we audited our Critical Suppliers and Economic Operators against MDR requirements?
CONCLUSION
The EU MDR does not just ask for a Quality Management System; it asks for a Total Lifecycle Management System. The goal is no longer just “consistency” (the ISO goal), but “safety and performance” (the MDR goal). By building these regulatory pillars into your QMS today, you ensure market access for tomorrow.
HOW MORULAA CAN HELP
At Morulaa, we specialize in supporting medical device manufacturers with end-to-end EU MDR compliance. From building or adapting your Quality Management System (QMS) to aligning it with Article 10 and Annex IX requirements, we offer tailored consulting, documentation templates, and audit readiness support. Whether you’re starting fresh or upgrading from MDD to MDR, we help ensure your QMS is not just compliant but effective.