

INTRODUCTION: THE MDR'S CONTINUOUS CHECK-UP
You’ve successfully brought a medical device to market. Great! But in the world of the EU Medical Device Regulation (MDR), getting the CE Mark is just the start. The real test begins when your device is actually being used by patients and healthcare professionals. This is where Post-Market Surveillance (PMS) comes in.
Think of PMS as your device’s compulsory, lifelong check-up. It’s not about waiting for a problem; it’s about proactively monitoring performance in the real world to ensure your device remains safe and effective over its entire lifespan.
The MDR makes this a mandatory, systematic process, making it a cornerstone of compliance and patient safety.
Check out our latest blog to learn more for
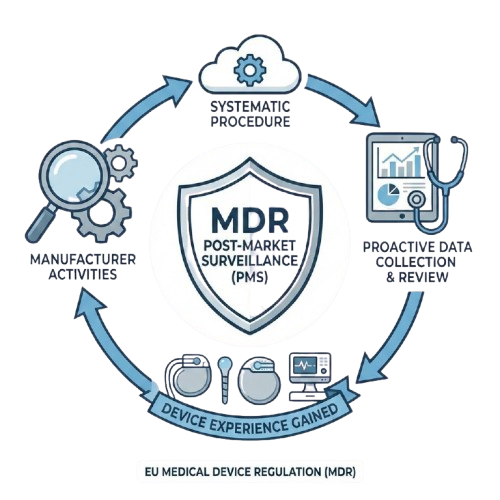
WHAT IS POST-MARKET SURVEILLANCE?
The MDR defines Post-Market Surveillance as all the activities a manufacturer carries out to establish and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market.
The goal of a robust PMS system is twofold:
- Continuous Improvement: The collected data is used to continually update the device’s technical documentation, clinical evaluation, and risk management system.
Safety First: It allows manufacturers to quickly identify the need for any preventive or corrective actions (including Field Safety Corrective Actions, or FSCAs) to improve the usability, performance, and safety of the device
Check out our latest blog to learn more for MDCG 2025-10 Published: The New Standard for Proactive Post-Market Surveillance
THE BLUEPRINT : YOUR POST-MARKET SURVEILLANCE PLAN
Every manufacturer must establish, document, and maintain a Post-Market Surveillance System based on a detailed Post-Market Surveillance Plan (PMSP). This plan is a required part of your technical documentation (Annex III of the MDR).
What Goes into the Plan?
The PMSP is not just a form; it’s a strategic process. It must be proactive and systematic and address the collection of all available information, including:
- Vigilance Data: Information concerning serious incidents and Field Safety Corrective Actions (FSCAs).
- Non-Serious Incidents and Side-Effects: Records referring to non-serious incidents and data on any undesirable side-effects.
- Trend Reports: Information from monitoring any statistically significant increase in the frequency or severity of non-serious incidents (Article 88).
- Feedback & Complaints: Information, including feedback and complaints, provided directly by users, distributors, and importers.
- Public Data: Relevant specialist or technical literature, databases, registers, and publicly available information about similar medical devices.
THE REPORTING ENGINE: PMSR VS. PSUR
The output of your PMS activities is summarised in mandatory safety reports, with the type of report depending on your device’s risk class:
Device Risk Class | Required Report | Description |
Class I (Lowest Risk) | Post-Market Surveillance Report (PMSR) | Summarises the results and conclusions of the PMS data analysis, including a rationale and description of any preventive and corrective actions taken. This report is updated as necessary and must be made available to the competent authority upon request. |
Class IIa, IIb, and III (Higher Risk) | Periodic Safety Update Report (PSUR) | This is a more comprehensive and formal report. It presents the results and conclusions of PMS data, with a focus on benefit-risk determination. The PSUR must be submitted to the Notified Body and is collated in the central European database on medical devices (Eudamed). |
POST-MARKET CLINICAL FOLLOW-UP (PMCF)
For many devices, especially higher-risk classes, PMS includes a key component: Post-Market Clinical Follow-up (PMCF).
PMCF is a continuous process that updates the device’s clinical evaluation. It involves proactively collecting and evaluating clinical data from the use of a CE-marked device to confirm its safety and performance over time, especially focusing on long-term risks or emerging issues. It’s the final piece of the puzzle that ensures the clinical evidence gathered before market entry holds true throughout the device’s life.
CONCLUSION
Under the EU MDR, Post-Market Surveillance is far more than just handling complaints. It is a proactive, required system that continuously feeds real-world data back into the design, risk management, and clinical evaluation of your device. A robust PMS system isn’t a regulatory burden, it’s the strongest evidence you have to demonstrate your commitment to long-term patient safety and product quality.
HOW MORULAA CAN HELP
At Morulaa, we support manufacturers with end-to-end Post-Market Surveillance (PMS) compliance under the EU MDR. We prepare fully compliant PMS Plans, PMSRs, PSURs, PMCF Plans, and PMCF Evaluation Reports tailored to your device and risk class. Our team ensures that your real-world data, vigilance inputs, complaint handling records, and clinical evidence flow seamlessly into your technical documentation.