
|
CE MARKING: YOUR PASSPORT TO THE EU
|
|---|
|
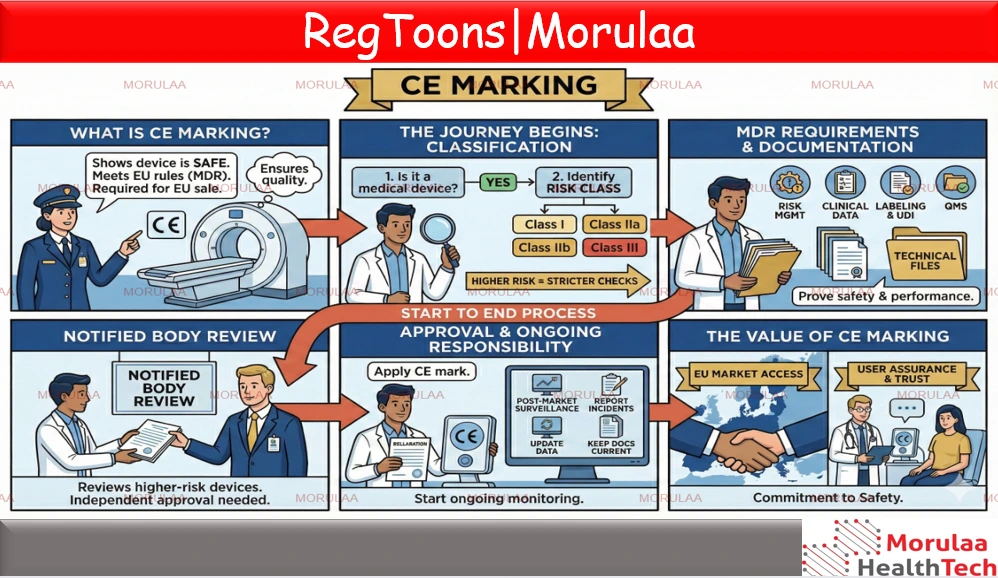
CE marking is a label that shows a medical device is safe to use and meets all rules required in the European Union. Under the EU Medical Device Regulation (MDR 2017/745), the CE mark means the manufacturer has checked the device carefully, followed strict safety and performance rules, and has proper documentation to prove it. Without CE marking, a medical device cannot be sold in the EU. To get CE marking, a manufacturer must first confirm that the product is a medical device and identify its risk class (Class I, IIa, IIb, or III). Higher-risk devices need stricter checks. Next, the manufacturer must follow MDR requirements that include creating a risk management file, clinical evaluation, labeling, UDI, and having a quality management system. All technical documents, design files, and clinical evidence must be compiled to show the device is safe. If the device is not low-risk, a Notified Body reviews and approves these documents before the CE mark can be used. After approval, the manufacturer issues a Declaration of Conformity and places the CE mark on the device. But the responsibility does not end there. Even after the product enters the market, the company must continue monitoring its safety through post-market surveillance, report incidents, update clinical data, and keep the technical documentation current. Overall, CE marking is not just a stamp it represents a commitment to safety, performance, and constant monitoring. It gives companies access to the EU market and assures users that the device meets high-quality and regulatory standards. |
INTRODUCTION
Medical devices sold in the European Economic Area (EEA) must carry CE marking, a symbol that signifies compliance with EU laws. Under Regulation (EU) 2017/745 (commonly referred to as MDR), CE marking serves as a manufacturer’s formal declaration that their device meets all applicable safety, performance, and quality requirements. It not only assures product safety but also enables the free movement of medical devices throughout the EU. The CE mark represents conformity with general obligations under the regulation, including risk management, clinical evaluation, and technical documentation. With the MDR’s emphasis on transparency and traceability, CE marking is more than just a label; it’s a continuous regulatory commitment.
WHY CE MARKING CHANGED: MDR VS MDD
Previously, CE marking was governed under the Medical Device Directive (MDD). While the MDD provided a framework, it lacked robustness in post-market oversight and clinical evaluation. Regulation (EU) 2017/745 introduced key updates, such as increased focus on clinical evidence, tighter scrutiny of Notified Bodies, and mandatory post-market surveillance systems. Unlike the MDD, which allowed for broader interpretation and had limited scope, the MDR provides a more comprehensive and enforceable legal framework. MDR also expands the definition of a medical device to include some aesthetic products and ensures consistent implementation across all EU Member States. The reform was driven by safety concerns and technological advances, necessitating a higher standard of vigilance and patient protection.
STEP-BY-STEP: HOW TO GET CE MARKING UNDER MDR
Step 1: Confirm Device Qualification and Classification
The first step is to verify whether your product qualifies as a medical device under Article 2 of the MDR. If it does, the next task is to determine its risk class (Class I, IIa, IIb, or III) based on its intended purpose and inherent risks, as outlined in Annex VIII. Classification affects every subsequent step, including the need for a Notified Body. High-risk devices require rigorous assessment procedures, while low-risk ones may follow self-declaration routes. A clear understanding of classification rules ensures the correct regulatory pathway and avoids delays in approval.
Step 2: Identify the Applicable Legal Requirements
After classification, identify all regulatory requirements your device must comply with. This includes the General Safety and Performance Requirements (Annex I), labeling and language provisions, clinical evaluation (Article 61), UDI requirements (Article 27), and the appropriate conformity assessment route (Article 52). Depending on the device’s features, other EU legislation may apply, such as REACH, RoHS, or regulations on electromagnetic compatibility. Compliance must be holistic and should consider both product and process requirements.
Step 3: Implement a Quality Management System (QMS)
A robust QMS is fundamental to CE marking. As mandated in Article 10, manufacturers must have a system in place that ensures continuous compliance throughout the device lifecycle. The QMS must cover production control, clinical evaluation, PMS, complaint handling, and corrective actions. Additionally, the manufacturer must appoint a Person Responsible for Regulatory Compliance (PRRC) who meets the qualification criteria specified in Article 15. For devices requiring Notified Body oversight, QMS certification is a prerequisite.
Step 4: Compile Technical Documentation
Technical documentation is the evidence that supports your conformity claim. As outlined in Annexes II and III, it must include device description, intended purpose, design specifications, risk management results, and clinical evidence. The documentation should also provide manufacturing details, labeling samples, UDI codes, and PMS/PMCF plans. It must be readily available for inspection by Notified Bodies or Competent Authorities and should be updated regularly to reflect design or regulatory changes.
Step 5: Conduct a Clinical Evaluation
Clinical evaluation is a systematic process to assess and verify a device’s safety and performance. According to Article 61 and Annex XIV, all devices must undergo this process, which can include clinical investigations, literature reviews, or equivalence data. For Class III and implantable devices, clinical investigations are usually required unless exemptions apply. The clinical evaluation must be thorough, evidence-based, and continuously updated via PMCF activities.
Step 6: Undergo Conformity Assessment
The conformity assessment route depends on the device’s risk classification. Class I devices may follow a self-assessment process, while Classes IIa, IIb, and III require Notified Body involvement. The assessment includes QMS audits, technical documentation reviews, and possibly product testing. Relevant conformity routes include Annex IX (Full QMS), Annex X (type examination), and Annex XI (product conformity verification). The objective is to verify compliance with all MDR requirements before market entry.
Step 7: Issue the EU Declaration of Conformity
Upon successful conformity assessment, the manufacturer must draft an EU Declaration of Conformity (Annex IV). This declaration is a legal document stating that the device meets all applicable MDR requirements. It must include details like the manufacturer’s information, UDI-DI, applicable standards or CS, and references to conformity assessment procedures. The declaration must be signed and kept up to date, and it should be available to Competent Authorities upon request.
Step 8: Affix the CE Marking
Finally, the CE mark must be affixed to the device, its sterile packaging, and any user instructions, following Article 20 and Annex V. If a Notified Body was involved, its four-digit identification number must accompany the CE mark. The marking must be clear, legible, and durable. It signifies that the device complies with MDR and can be freely sold in the EU. Improper use of the CE mark is prohibited and subject to enforcement action.
RESPONSIBILITIES AFTER CE MARKING
Post-Market Surveillance (PMS)
Manufacturers are obligated to monitor their devices once placed on the market. PMS includes the collection, analysis, and use of real-world performance data to identify any need for corrective or preventive actions. Article 83 requires that PMS systems be part of the QMS and proportionate to the device’s risk. PMS findings must inform risk management, labeling updates, and clinical evaluation revisions.
Post-Market Clinical Follow-Up (PMCF)
PMCF is an ongoing process that complements the clinical evaluation. It involves actively collecting clinical data from post-market use to confirm safety and performance over time. As part of Annex XIV Part B, PMCF is mandatory for most devices, especially those that are high-risk. It helps detect rare complications, long-term effects, or misuse issues that may not be evident during pre-market studies.
Vigilance & Incident Reporting
Manufacturers must report serious incidents, device deficiencies, and Field Safety Corrective Actions (FSCAs) to Competent Authorities within strict timelines. Articles 87 to 89 outline the vigilance requirements, including 2-day reporting for serious threats and 10-day limits for other reportable events. Timely reporting ensures patient safety and regulatory compliance, and it allows authorities to coordinate action across Member States.
Labeling and UDI
Proper labeling is essential for traceability and user safety. MDR requires that labels include the UDI, manufacturer details, warnings, and symbols as per Annex I, Section 23. Devices must be labeled in the official language(s) of the target country. UDI, introduced under Article 27 and Annex VI, enables device tracking throughout the supply chain and helps reduce errors and counterfeit risks.
WHAT DOES THE CE MARK REALLY MEAN?
The CE mark is not just a logo, it’s a declaration of regulatory compliance. It means the manufacturer has met all applicable requirements of MDR 2017/745, including design controls, risk management, clinical evaluation, labeling, and post-market obligations. The mark gives access to the EU market and signifies a commitment to safety and performance throughout the device’s lifecycle.
Examples of CE Mark Use under MDR
- Class I Devices: Products like surgical gloves and bandages, where manufacturers can self-declare conformity under MDR requirements.
- Class IIa Devices: Devices such as dental fillings, which must undergo assessment and approval from a Notified Body.
- Class III Devices: High‑risk devices like heart valves that require comprehensive clinical investigations along with an in‑depth review by a Notified Body.
CONCLUSION
Obtaining CE marking is a structured process that involves legal, technical, and quality assurance disciplines. With MDR in force, the bar for compliance is higher than ever. Manufacturers must invest in strong internal systems and understand the expectations at each step. CE marking is not a one-time task but an ongoing responsibility that requires vigilance, transparency, and dedication to patient safety.
HOW MORULAA CAN HELP
Morulaa HealthTech helps global medical device manufacturers navigate complex regulatory landscapes by providing expert support for product registration, importation, and post-market compliance in India and other key markets. With a unique independent license holder model, Morulaa enables companies to collaborate with multiple distributors without duplicating registrations, ensuring faster market access, regulatory clarity, and strategic growth.