
Overview of Process Flow Rapid Alert & Recall System
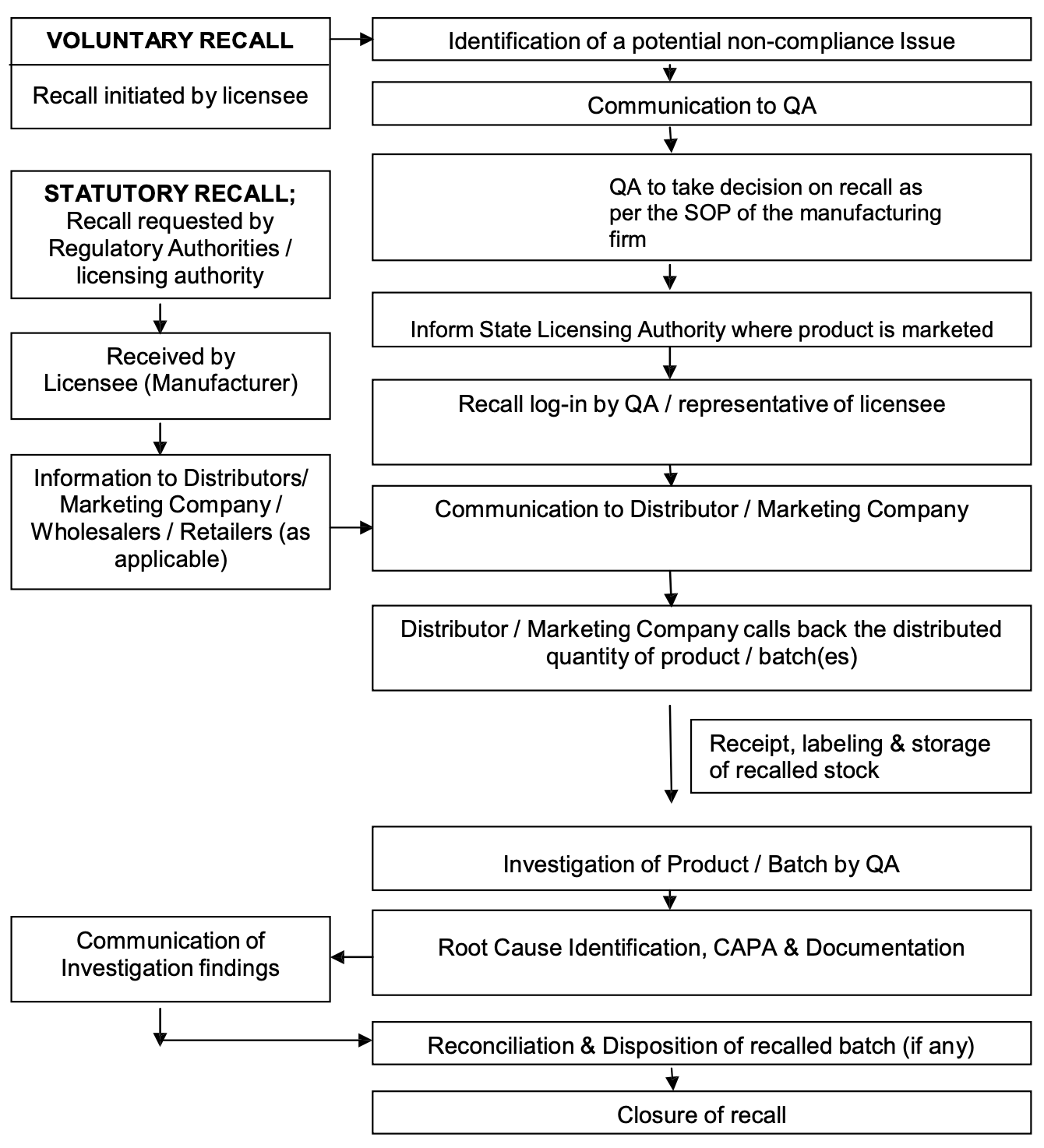
Procedure for Recall
The foundation of a secure and effective recall system lies in the preparedness and organization of the product manufacturers or importers. Key elements of this strategy include:
-
Designation of Responsibility: An authorized individual must be appointed to oversee and coordinate recall efforts. This role involves ensuring that adequate staffing and resources are available to manage recalls efficiently and effectively.
-
Development of Written Procedures: Clear, written procedures for organizing and executing recall activities is important. These procedures should be regularly reviewed and updated, detailing how recalls can be initiated at the necessary level within the distribution chain.
-
Segregation of Recalled Products: Recalled items must be stored in a secure, segregated area to prevent their re-entry into the market.
-
Notification of Licensing Authorities: Regulatory bodies, such as the CDSCO in India, must be informed of any recall plans, highlighting the importance of transparency and communication with licensing authorities.
-
Accessible Distribution Records: Detailed records of distribution, including information on wholesalers and direct customers, must be easily accessible. This ensures that recalls can be carried out effectively, reaching all affected products in the distribution chain.
-
Monitoring and Documentation: The recall process must be closely monitored and documented, including the final disposition of the recalled product. A final report reconciling delivered and recovered product quantities is important for evaluating the recall’s effectiveness.