
|
WHEN DEVICES MEET RULES: MDR FOR THE REST OF US
|
|---|
|
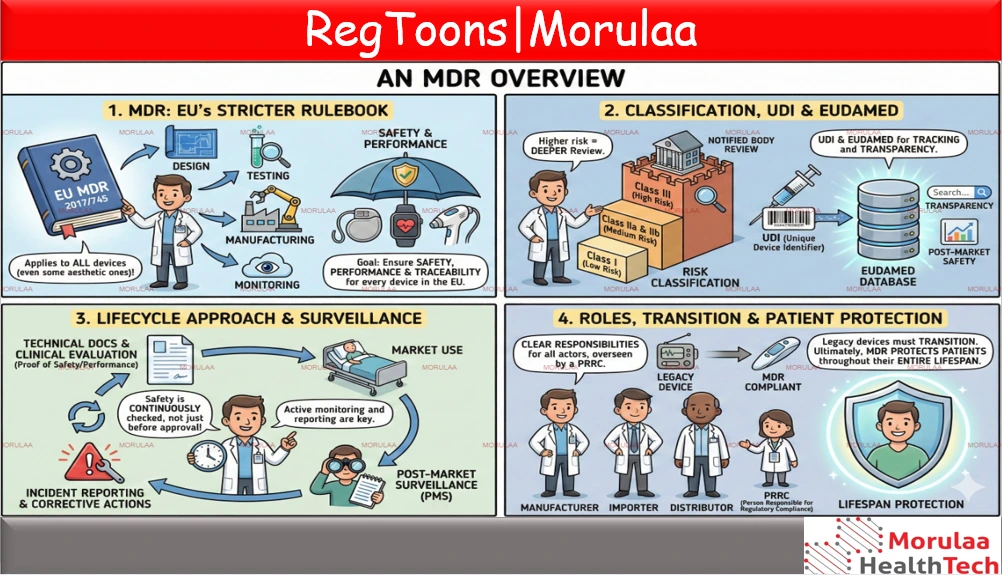
The EU Medical Device Regulation (MDR 2017/745) is the European Union’s updated and much stricter rulebook for how medical devices must be designed, tested, manufactured, and monitored. It applies to all devices used on humans, including some that do not have a medical purpose (like certain cosmetic or aesthetic devices). The goal is to ensure safety, performance, and full traceability of every device placed on the EU market. Under the MDR, manufacturers must classify their devices by risk (Class I, IIa, IIb, III) and meet the requirements for that category; higher-risk devices must undergo deeper review by a Notified Body. Every device must also have a UDI (Unique Device Identifier) to allow tracking through the supply chain, and device information must be registered in the EU database, EUDAMED, to support transparency and post-market safety monitoring. A key part of MDR is strong technical documentation, proof of safety and performance, and ongoing clinical evaluation using real-world or study data. Once a device is on the market, manufacturers must actively monitor it through post-market surveillance (PMS), report incidents, and take corrective actions when needed. This “lifecycle” approach ensures that safety is continuously checked, not only before approval. The MDR also clearly defines the responsibilities of manufacturers, authorised representatives, importers and distributors, and requires a Person Responsible for Regulatory Compliance (PRRC) to oversee conformity. For older (“legacy”) devices, the MDR includes transitional rules, meaning manufacturers must eventually upgrade them to MDR requirements. Overall, MDR is designed to protect patients by making sure devices are safe, high quality, and continuously monitored throughout their entire lifespan. |
INTRODUCTION
The Regulation (EU) 2017/745 represents a sweeping reform of medical‑device regulation in the European Union. It places rigorous demands on manufacturers, importers, distributors, notified bodies and other economic operators to ensure patient and user safety, device performance and traceability across the lifecycle. Below is a section‑by‑section breakdown of the main regulatory topics, with explanations of what each means for manufacturers like you (or “us”, in line with your preferred phrasing).
SCOPE OF THE EU MDR REGULATIONS 2017/745
What the section covers
The MDR sets out its application and definitions in Chapter I (Articles 1 to 4). It covers medical devices and their accessories intended for human use, devices for diagnosis/monitoring/prevention/treatment/compensation of disease or disability, and also devices without a medical purpose listed in Annex XVI. It defines key terms such as “manufacturer”, “placing on the market”, “making available on the market”, “device”, “accessory”, etc.
What it means for manufacturers
- We must first confirm whether our product falls under the MDR’s scope — if so, all the subsequent requirements apply.
- If our device is intended for human use and meets the definition of a “medical device”, then the full regime applies.
- If the device is non‑medical but listed in Annex XVI (for example certain aesthetic devices), the MDR may still apply.
- We must clearly understand definitions (e.g., what is “placing on the market”) because these determine when compliance obligations start.
- Even if the device was previously placed under the older directive (Directive 93/42/EEC or Directive 90/385/EEC), the MDR’s transitional and excluding rules (Chapter X) may apply.
CLASSIFICATION OF DEVICES
What the section covers
Device Class | Risk Level | Regulatory Oversight | Typical Devices |
Class I | Low | Self-certification (except if sterile/measuring) | Bandages, Stethoscopes |
Class IIa | Medium | Notified Body involvement required | Hearing aids, dental fillings |
Class IIb | Medium to High | Higher Notified Body involvement | Ventilators, infusion pumps |
Class III | High | Full Notified Body scrutiny | Heart valves, implantable pacemakers |
What it means for manufacturers
- We must determine and document the correct risk class for our device using the rules in Annex VIII.
- The class determines the level of regulatory oversight: higher‑risk devices (IIb, III) typically involve a notified body, more stringent clinical and technical requirements.
- Mis‑classifying the device (or failing to justify the classification) may lead to non‑compliance, market withdrawal or regulatory actions.
- For devices already on the market under older regimes, we must review any reclassification under MDR, especially where rules have change
UNIQUE DEVICE IDENTIFICATION (UDI) & TRACEABILITY
What the section covers
Chapter III of the MDR deals with device identification and traceability, including the UDI system. Annex VI covers UDI and EUDAMED registration. The UDI system requires each device (or grouping) to carry a unique identifier facilitating traceability and post‑market surveillance.
What it means for manufacturers
- We must assign a UDI‑DI (device identifier) and UDI‑PI (production identifier) according to the MDR rules.
- We need to register device information in the EU database for medical devices (EUDAMED) and maintain the data (e.g., device status, packaging changes).
- Labeling and packaging must include the UDI as required.
- Traceability improves our ability to conduct recalls/corrections efficiently and supports post‑market monitoring and vigilance.
- We should integrate UDI planning early in the device lifecycle (design, manufacturing, labeling) to avoid delays later.
TECHNICAL DOCUMENTATION & GENERAL SAFETY AND PERFORMANCE REQUIREMENTS (GSPR)
What the section covers
Chapter II (Articles 10 to 15) prescribes the obligations for manufacturers, and Annex I lists the General Safety and Performance Requirements (GSPR) that devices must satisfy. Annex II sets out the contents of the technical documentation. The technical documentation must demonstrate conformity with the GSPR and the relevant harmonised standards or common specifications.
What it means for manufacturers
- We must prepare, maintain and update sufficient technical documentation (the “technical file”) for each device or device family. This includes device description, design and manufacturing information, risk management (e.g., per ISO 14971), verification & validation data, clinical data, labeling, instructions for use, etc.
- We must ensure the device meets all applicable GSPR in Annex I – this means safety (e.g., biocompatibility, mechanical safety, electrical safety, software reliability) and performance (i.e., the device does what it’s intended to do).
- Where applicable, we must reference harmonised standards (e.g., ISO/IEC, EN standards) or common specifications if no standard exists. Compliance with harmonised standards provides a presumption of conformity.
- The technical documentation must be readily available for authorities and must be kept for a certain period (e.g., 10 years after the last device is placed on the market; for implantables possibly 15 years).
- Any changes in the device (design, manufacturing site, key components) must prompt updates to the technical documentation and possibly a new conformity assessment (depending on class of device and nature of change).
CLINICAL EVALUATION & CLINICAL INVESTIGATIONS
What the section covers
Chapter VI (Articles 61 to 82) covers clinical evaluation and investigation. The MDR requires that manufacturers perform a clinical evaluation to confirm the device’s safety and performance, relying on clinical data, literature, post‑market experience or new clinical investigations when necessary. Annex XIV further sets out detailed requirements for clinical investigations.
What it means for manufacturers
- We must perform a clinical evaluation per the requirements: collect existing clinical data (literature, previous device experience), analyse and justify its adequacy, and where there are gaps, plan and conduct a clinical investigation.
- For high‑risk devices (Class III, implantables) typically clinical investigations are required unless a justified exemption applies.
- Clinical evaluation is not a one‑off: it must be kept up‑to‑date (“lifecycle”) — we must monitor continuing safety and performance via PMS (post‑market) data and update evaluations accordingly.
- Documentation of the clinical evaluation (report) must be part of the technical file and be available for competent authorities.
- Investigational devices must meet specific protocols, ethics approvals, patient protections, and after completion, results must feed into the evaluation and risk/benefit analysis.
- Ensuring alignment with regulatory expectations (including by national competent authorities and via the Medical Device Coordination Group guidance) avoids delays or rework.
CONFORMITY ASSESSMENT & CE MARKING
What the section covers
Chapter V (Articles 52 to 60) covers classification and conformity assessment procedures. Depending on the device class, different routes apply from self‑certification (for low risk) to fully notified‑body audits, product verification, type examination, etc. Once conformity is demonstrated, a Declaration of Conformity is issued and the CE mark affixed. Annexes IX–XI lay out detailed procedures.
What it means for manufacturers
- We must select and follow the appropriate conformity assessment procedure for our device class. For example, Class I (non‑sterile, non‑measuring) may allow self‑certification (if no measuring function & no sterile packaging) but still must meet GSPR and technical documentation.
- For higher‑risk classes, we must engage a notified body (NB) which audits QMS, reviews technical documentation, may conduct audits of manufacturing and supply chain, and issue a certificate.
- After successful assessment, we prepare the EU Declaration of Conformity, affix the CE mark, and place the device on the market.
- We must ensure that any changes to the device, manufacturing process, or intended use are evaluated for their impact on conformity – substantial changes may require new assessment.
- Proper vigilance and PMS data are relevant not only post‑market but also feed into conformity justification over device lifecycle.
POST‑MARKET SURVEILLANCE (PMS), VIGILANCE & MARKET SURVEILLANCE
What the section covers
Chapter VII (Articles 83 to 100) covers post‑market surveillance by manufacturers, market surveillance by Member States, vigilance and reporting of serious incidents or safety corrective actions. Annex III sets documentation for PMS; Annex XIII deals with custom‑made devices.
What it means for manufacturers:
- We must establish a PMS system and implement a PMS plan for each device or device family. The PMS system must collect and analyse data about the device once it’s placed on the market, identify trends, and feed this into risk‑management and review processes.
- We must prepare a Periodic Safety Update Report (PSUR) for Class IIa and higher devices and other summary reports as required.
- When a serious incident occurs or a field safety corrective action (FSCA) is needed, we must report to the competent authority without undue delay.
- We must implement corrective or preventive actions (CAPA) when trends or data reveal increased risks or decreased performance. We must also maintain documentation (PMS data, reports) and external communication (e.g., via EUDAMED).
- Market surveillance authorities may inspect documents, access technical files, review PMS data, check UDI/traceability, and enforce actions if the device is non‑compliant or unsafe.
- The PMS loop is continuous — from design inputs, clinical data, manufacturing, to market feedback and back.
- We must establish a PMS system and implement a PMS plan for each device or device family. The PMS system must collect and analyse data about the device once it’s placed on the market, identify trends, and feed this into risk‑management and review processes.
ECONOMIC OPERATORS AND ROLES (MANUFACTURER, AUTHORISED REPRESENTATIVE, IMPORTER, DISTRIBUTOR)
What the section covers
Chapter II (especially Articles 10 to 15) and elsewhere define obligations of different economic operators: manufacturers, authorised representatives (for non‑EU manufacturers), importers, distributors, and other actors.
What it means for manufacturers (and us):
- As a manufacturer, we hold primary responsibility for device conformity, technical documentation, QMS, PMS, vigilance, labeling, instructions for use, providing necessary information to importers/distributors, and registration with markets.
- If we are a non‑EU manufacturer placing devices via an authorised representative in the EU, that representative will share liability and must be designated in writing.
- Importers and distributors must verify CE marking, correct labeling, registration in EUDAMED, UDI information, and must have systems to verify compliance and maintain traceability.
- We must ensure that our supply chain (including distributors and importers) understands their roles and that we maintain adequate agreements and oversight.
- We must ensure records of economic operators (traceability) for devices we place on the market.
PERSON RESPONSIBLE FOR REGULATORY COMPLIANCE (PRRC)
What the section covers:
Article 15 requires every manufacturer (and authorised representative) to have at least one Person Responsible for Regulatory Compliance (PRRC) who meets certain qualifications and ensures conformity obligations are met.
What it means for manufacturers:
- We must designate a PRRC who has scientific/regulatory background or equivalent experience and ensures that device conformity, technical documentation, QMS, PMS and vigilance obligations are fulfilled.
- The PRRC must be permanently available within the organisation (or via contractual arrangement) and must sign off on device conformity, documentation and obligations.
- This adds organisational accountability: the PRRC is a named individual who can be approached by competent authorities regarding compliance and documentation.
- We must ensure that the PRRC’s responsibilities are clearly described, training is provided, and that internal processes support them (e.g., document management, CAPA, audits).
EUDAMED & PUBLIC TRANSPARENCY
What the section covers
Chapter III (Traceability) and other chapters refer to the EU database for medical devices (EUDAMED). The database supports registration, UDI/traceability, vigilance, clinical investigations, certificates of notified bodies, and more.
What it means for manufacturers:
- We must register our devices and related economic‑operator data in EUDAMED when the modules become fully operational.
- We must upload relevant data (device registration, UDI‑DI/PI, vigilance reports, certificates, performance studies) to achieve transparency and fulfil traceability obligations.
- The public transparency aspect means certain device information becomes accessible to stakeholders (patients, clinicians, authorities) – so we must ensure data accuracy and timeliness.
- We must keep records up to date (e.g., device status, changes, recalls) and monitor modules in EUDAMED for our obligations.
TRANSITIONAL PROVISIONS & LEGACY DEVICES
What the section covers:
Chapter X (Articles 97 to 105) includes transitional provisions for devices previously placed under the older directives and devices already on the market, along with dates for full application of the MDR.
What it means for manufacturers:
- If we have devices currently on the market under the older directive (MDD or AIMDD), we must assess whether they can transition to MDR under the grandfathering rules, or whether we must re-certify under MDR to continue placing them on the market.
- We must monitor official corrigenda, amendments and extensions of transitional periods (for example extensions made in March 2023) so we don’t miss compliance deadlines.
- We must plan for devices whose certificates expire or are re-issued under MDR, gaps in supply, or changes in notified‑body availability due to the MDR administration.
CUSTOM‑MADE DEVICES & DEVICES WITHOUT INTENDED MEDICAL PURPOSE
What the section covers:
Annex XIII covers custom‑made devices, and Annex XVI covers devices without an intended medical purpose. The MDR provides special rules for these categories.
What it means for manufacturers:
- If we produce custom‑made devices (for a specific patient, made in response to a prescription), we still must meet certain obligations (technical documentation, PMS, labeling), even though the conformity assessment route may be simplified.
- If we produce devices without intended medical purpose (listed in Annex XVI), we must determine whether the MDR applies and ensure we treat them according to the special rules (including classification, documentation, performance requirements) as though they were medical devices.
- We must clearly identify the device category, justify any exemptions, and ensure full traceability and documentation even for these special categories.
MARKET SURVEILLANCE, ENFORCEMENT, PENALTIES
What the section covers
Chapter IX (Articles 102 to 105) addresses confidentiality, data protection, penalties, coordination and cooperation between competent authorities and the Medical Device Coordination Group (MDCG).
What it means for manufacturers
- We should anticipate market‑surveillance audits by national competent authorities. This includes inspection of technical documentation, QMS records, PMS data, UDI/traceability, etc.
- Non‑compliance may lead to penalties, device withdrawal, recall, suspension of CE‐marking, or prohibition orders.
- We must maintain proper documentation, ensure we retain records for required retention periods, respond to authorities promptly, and maintain transparency with regulatory bodies.
- We must monitor the regulatory landscape (via MDCG guidance, corrigenda, updates) and update our compliance processes accordingly.
QUALITY MANAGEMENT SYSTEM (QMS) & RISK MANAGEMENT
What the section covers
While not a separate heading in the regulation, Chapter II and Annexes refer to manufacturers’ obligation to implement a QMS (including for design, manufacturing, distribution) and risk management across the device lifecycle.
What it means for manufacturers
- We must establish and maintain a QMS (often aligned with ISO 13485) for design and manufacture of devices in scope of the MDR (except certain Class I devices which may use simplified QMS). The QMS must include processes for design control, production, service, post‑market activities, CAPA, internal audits, management review, etc.
- Risk management per ISO 14971 must be applied throughout the device lifecycle: design, manufacture, distribution, post‑market, field‑use, retirement. We must demonstrate that benefits outweigh residual risks, and we must monitor and reassess risks if new data emerges.
- The QMS and risk‑management documentation will be assessed by notified bodies (or competent authorities) as part of conformity assessment, so we must ensure records are robust, up to date, and accessible.
Check out our latest detailed blog post : Risk management system and Quality management system
CONCLUSION
The MDR fundamentally raises for medical‑device regulation within the EU, emphasising full‑lifecycle oversight from design, manufacture, clinical evaluation to post‑market surveillance and traceability. For us as manufacturers (or service providers working on behalf of them), compliance means more than ticking boxes: it means embedding regulatory strategy early, ensuring strong documentation, maintaining traceability, proactively managing risk and post‑market feedback, and staying alongside evolving regulatory guidance.
HOW MORULAA CAN HELP
Morulaa simplifies your MDR 2017/745 compliance journey by acting as your dedicated regulatory partner. From classifying your medical devices correctly, preparing robust technical documentation, managing clinical evaluation and PMS plans, to serving as an EU Authorized Representative if needed — we help manufacturers meet every critical MDR requirement efficiently. With deep regulatory expertise and end-to-end support, we ensure your devices are audit-ready and market compliant, so you can focus on innovation while we handle compliance.